
· are interested in genetic testing for autism
·
are affected by miss-expression of the
genes:-
·
ANKRD17
·
TCF4
·
CNTNAP2
·
NRXN1
It is one of those posts that could go on forever; the more you dig, the more you uncover and you wonder why other people (salaried researchers) are not doing this.
Today’s post is mainly about a gene called ANKRD17, but it does highlight more general
principles about those genetic testing results, some parents strive to
obtain. It does look at downstream
effects of Pitt Hopkins Syndrome and “Pitt Hopkins-like Syndrome”, which likely merge into mainstream autism.
Many single gene autisms have already been identified, some have names and some are still SWANs (Syndromes Without A Name). Some syndromes have long been identified, but their biological basis had not been identified. From last year, loss of function of the gene ANKRD17 became Chopra-Amiel-Gordon Syndrome.
Our reader Mary’s whole exome sequencing (WEG) from a few
years ago, for her daughter, has now been flagged up as carrying a mutation
leading to Chopra-Amiel-Gordon Syndrome.
In effect, the mutation in ANKRD17 went from be of no confirmed relevance to autism,
to being causal, thanks to Dr Chopra and her pals.
This highlights the weakness in the
interpretation of genetic testing. Any
benchmark list of autism genes is just a work in progress; your mutation may
not yet be there.
Another gene recently queried by a
reader was CNTNAP2,
this turns out to a key DEG (differentially expressed gene) of a syndrome with
its own name, Pitt Hopkins Syndrome, caused by reduced expression of TCF4
(Transcription Factor 4). Reduced
expression of TCF4 has very many effects, but one effect is to reduce expression of
CNTNAP2.
In lay-speak, lack of TCF4 causes a cascade of effects, one of which is on the expression of CNTAP2. We see that people with a CNTAP2 mutation share many of the features of people having a TCF4 mutation. So, of all the many effects caused by TCF4, those along the TCF4-CNTAP2 pathway should be focused on. The mutation in CNTNAP2, quite rationally, is now called Pitt Hopkin-like Syndrome-1. There is also a Pitt Hopkin-like Syndrome-2 which is caused by a mutation in NRXN1 (neurexin 1).
https://royalsocietypublishing.org/doi/pdf/10.1098/rsob.210091
In mammals, the neurexins are
encoded by three NRXN genes (NRXN1-3), each of which has both an upstream
promoter that is used to generate the α-neurexins, and a downstream promoter
that is used to generate the shorter β-neurexins [13,15].
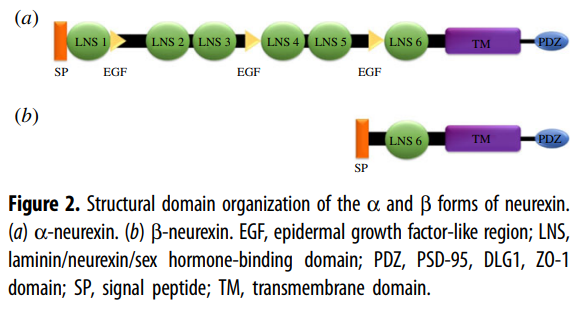
α-neurexins are composed of six large extracellular
laminin/neurexin/sex hormone-binding (LNS) globulin domains with three
interspersed epidermal growth factor (EGF)-like regions
Just note the term EGF.
In very recent research we see that a reduction in epithelial growth factor may be
what is driving some of the key clinical features, such as lack of language.
CNTNAP2 has been identified as a master gene in autism manifestation responsible for speech-language delay by impairing the EGF protein domain and downstream cascade. The decrease in EGF is correlated with vital autism symptoms, especially language disabilities.
Autism exhibits genetic heterogeneity, and
hence, it becomes difficult to pinpoint one single gene for its manifestation.
The gene clusters with varied pathways show the convergence of multiple gene
variants, resulting in autism manifestation. Whole-exome sequencing proves to
be a reliable tool for deciphering the causal genes for autism manifestation.
Deciphering the autism exome identified the mutational landscape derived from
single and multi-base DNA variants. Genes carrying mutations were identified in
synaptogenesis processes, EGF signaling, and PI3K/MAPK signaling. Protein-protein interactions of
NrCAM and CNTN4 with CNTNAP2 increased the impact and burden on autism.
Shining a light on CNTNAP2: complex
functions to complex disorders
TCF4 encodes a basic helix-loop-helix (bHLH) transcription factor that binds near the start site of CNTNAP2 to upregulate its expression (Figure 1a).48 In humans, TCF4 is more highly expressed in the neocortex and hippocampus than in the striatum, thalamus and cerebellum.49 Mutations in TCF4 have been shown to cause Pitt–Hopkins syndrome (PTHS) and three rare TCF4 SNPs are associated with schizophrenia.17, 49, 50, 51 PTHS is characterised by severe intellectual disability, absent or severely impaired speech, characteristic facial features and epilepsy.52 Many of these features are shared with patients carrying CNTNAP2 mutations, leading researchers to test patients with PTHS-like features for CNTNAP2 mutations.17 Two mutations affecting the CNTNAP2 locus (one homozygous and one compound heterozygote) were identified in two independent pedigrees (Table 1). This suggested that disruption of the TCF4–CNTNAP2 pathway could be related to intellectual disability, seizures, and/or behavioural abnormalities.
One of our readers in Australia recently queried the potential
significance of a mutation (an SNP) in CNTNAP2.
Based on the above, it clearly could be very important.
What is the common link between TCF4, CNTNAP2 and NRXN1? It would
seem to be EGF (epidermal growth
factor).
It looks quite possible that EGF is
disturbed in much broader autism. It appears that inflammation may reduce EGF
levels. It is a rather circular argument, but we also know that EGF reduces
inflammation.
To sum up people, with autism likely want more EGF and we already knew that they definitely want less inflammation.
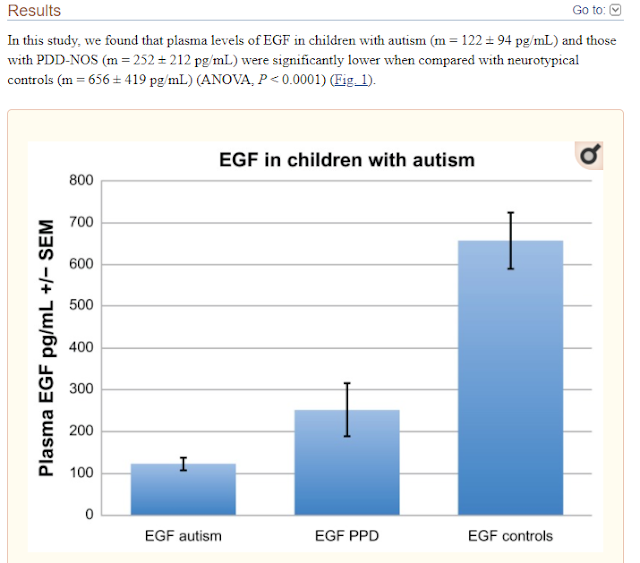
Decreased Epidermal Growth Factor (EGF) Associated with HMGB1 and Increased Hyperactivity in Children with Autism
These results suggest an association between
decreased plasma EGF levels and selected symptom severity. We also found a
strong correlation between plasma EGF and HMGB1, suggesting inflammation is
associated with decreased EGF.
ANKRD17
Finally, we get back to ANKRD17.
Our reader Mary has already highlighted
this recent paper: -
Heterozygous ANKRD17 loss-of-function variants cause a
syndrome with intellectual disability, speech delay, and dysmorphism

Dysmorphic facial features of the ANKRD17-related
disorder
ANKRD17 is an ankyrin repeat-containing
protein thought to play a role in cell cycle progression, whose ortholog
in Drosophila functions
in the Hippo pathway as a co-factor of Yorkie. Here, we delineate a
neurodevelopmental disorder caused by de
novo heterozygous ANKRD17 variants.
The mutational spectrum of this cohort of 34 individuals from 32 families is
highly suggestive of haploinsufficiency as the underlying mechanism
of disease, with 21 truncating or essential splice site variants, 9 missense
variants, 1 in-frame insertion-deletion, and 1 microdeletion (1.16 Mb).
Consequently, our data indicate that loss of ANKRD17 is
likely the main cause of phenotypes previously associated with large
multi-gene chromosomal aberrations of the 4q13.3 region. Protein
modeling suggests that most of the missense variants disrupt the stability
of the ankyrin repeats through alteration of core structural residues. The major phenotypic
characteristic of our cohort is a variable degree of developmental
delay/intellectual disability, particularly affecting speech, while additional
features include growth failure, feeding difficulties, non-specific MRI
abnormalities, epilepsy and/or abnormal EEG, predisposition to recurrent
infections (mostly bacterial), ophthalmological abnormalities, gait/balance
disturbance, and joint hypermobility. Moreover, many individuals shared similar
dysmorphic facial features. Analysis of single-cell RNA-seq data from
the developing human telencephalon indicated ANKRD17 expression
at multiple stages of neurogenesis, adding further evidence to the
assertion that damaging ANKRD17 variants cause a neurodevelopmental
disorder.
Neonatal growth parameters were normal in the majority of
individuals (Table S2) but postnatal
growth failure was a feature of almost half of the individuals (height
< 2 SD in n ¼ 12 and weight < 2 SD in n ¼ 9). One individual with marked
growth failure (individual 3, height 3.8 SD) was under treatment with growth
hormone (GH), although GH stimulation testing was normal. Feeding difficulties,
especially reduced oral intake, were reported at some stage in 11 individuals,
5 of whom required G-tube nutritional supplementation. Postnatal microcephaly (OFC < 2SD) was noted
in seven individuals, and macrocephaly in four (one of these individuals, however, also
harbored a pathogenic de novo NSD1 variant (GenBank: NM_022455.4, c.2615T>G
[p.Leu872*]). Epilepsy was
reported in nine individuals (individuals 1, 2, 16, 19, 21, 25, 27, 28,
and 33), with an age of onset of under 2 years for five individuals
(individuals 1, 2, 16, 19, and 25). Focal seizures with secondary
generalization was the most common seizure subtype, present in five individuals
(individuals 1, 2, 21, 25, and 27). One individual had Lennox-Gastaut epilepsy
(individual 16), one had tonic seizures with head deviation (individual 19),
one had mixed myoclonic and tonic-clonic epilepsy (individual 33), and another
a mixture of tonic-clonic and absence seizures (individual 28). Seizures were
well controlled (less frequent than every 2 years) in five individuals (individuals
2, 21, 25, 28, and 33), all of whom were on three or fewer antiepileptic drugs
(AEDs). Moderate control, with seizures every 2–3 months, was reported in
individual 1, who was on Valproate monotherapy. Two individuals had refractory
epilepsy during at least parts of their disease course—individual 19 who had
frequent tonic seizures in infancy that resolved with topiramate monotherapy
and individual 16 Table 2. Frequencies of phenotypic characteristics of
individuals with ANKRD17 variants Frequency Sex F ¼ 19, M ¼ 15 Growth Height
< 2 SD 12/31 Weight < 2 SD 9/30 OFC < 2 SD 7/31 OFC > 2 SD 4/31
Development DD or ID 31/34 severe 7 moderate 12 mild 5 borderline 7 Motor delay
20/29 Speech delaya 29/32 Other ASD, n ¼ 8; ADHD, n ¼ 4 Neurology Epilepsy 9/33
Abnormal EEG 10/23 Brain MRI abnormalities 11/23 Gait or balance abnormalities
9/25 Spasticity or hypertonia 4/26 Other Recurrent infections 11/33 Feeding problems 11/27 Palate
abnormalities 3/34 Hypermobility 9/29 Ophthalmological abnormalities 13/23 Miscellaneous
Minor digital anomalies 6 Genitourinary abnormalities 5 Pigmentary
abnormalities 4 Scoliosis 3 Abnormal bone mineralization 2 Prominent blood
vessels 2 ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum
disorder a For details see Table S1 The American Journal of Human Genetics 108,
1138–1150, June 3, 2021 1143 who had multiple seizures every day despite three
AEDs. Further details of epilepsy phenotype, including previously trialled
AEDs, are noted in Table S2. There were four individuals without epilepsy in
whom an abnormal EEG was recorded. Other neurological features include poor
balance and/or abnormal gait (9/25) and peripheral spasticity (4/26, one of
whom one was microcephalic). Neuroimaging
abnormalities were identified in 11 of the 23 individuals in whom an MRI was
recorded. Abnormalities include decreased white matter volume (individuals 14,
16, and 18), thinning of the corpus callosum (individuals 14 and 19), optic
nerve hypoplasia (individuals 18 and 19), a localized hyperintensity
(individuals 7 and 31), right temporal sclerosis (individual 16), dilated
Virchow-Robin spaces (individual 6), periventricular nodular heterotopia
(individual 30), and an arachnoid (individual 24) and pineal cyst (individual
16). Ophthalmological
abnormalities were reported in 13/23 individuals. There were nine individuals with
recurrent bacterial infections, one with recurrent viral infections, and
one individual with recurrent infections that were both viral and bacterial.
The source of bacterial infection was primarily the upper and lower respiratory
system and the middle ear (nine individuals) and in some cases required
hospitalization. Two individuals were on low-dose prophylactic antibiotics for
recurrent otitis media or respiratory tract infections. Notably, individual 26
had a history of pseudomonas and methicillin-resistant staphylococcal aureus
(MRSA) infection on his toes. Immunology assessments were recorded in five
individuals, details of which can be found in Table S2, with no obvious
immunodeficiency identified in these individuals. Generalized joint
hypermobility was reported in 9/29 individuals. Notably, there were two
individuals with cleft palate in the context of Pierre Robin sequence (PRS) and
another with cleft lip and palate. Other infrequent features include minor
digital anomalies (n ¼ 6), genitourinary abnormalities (n ¼ 5, of whom three
had unilateral renal agenesis), abnormal skin pigmentation (n ¼ 4), scoliosis
(n ¼ 3), abnormality of bone mineralization (n ¼ 2), and cutaneous prominence
of blood vessels (n ¼ 2). Figure 2 shows the facial features of individuals
with the ANKRD17-related neurodevelopmental disorder. Key dysmorphic features
include a triangular-shaped face found in 10 of the 24 individuals for whom photos
were available with a high anterior hairline (19/24), eyes which are either
deep-set (5/24) or almond shaped (8/24) with periorbital fullness (6/24), thick
nasal alae and flared nostrils (9/24), full cheeks (7/24), and a thin upper lip
(12/24). The degree of dysmorphism was variable, with several individuals
(particularly individuals 8 and 10) presenting with only subtle dysmorphic
characteristics. Persistence of the high anterior hairline, periorbital
fullness, and full cheeks into adulthood is demonstrated in individual 12 (age
30 years) and individual 25 (age 34 years). A number of diagnoses had been
considered in several individuals prior to the identification of an ANKRD17
variant, including SATB2-associated syndrome (MIM: 612313) in individual 5 who
presented with PRS, triangular facies and speech delay, and Floating-Harbour
syndrome (MIM: 136140) in individual 9 who presented with marked short stature
(height < 3 SD), microcephaly (head circumference < 2.5 SD), dysmorphic
features, and borderline ID. This
highlights the phenotypic overlap of the ANKRD17-related disorder with a number
of other genetic syndromes, notably those with expressive language delay. In
our cohort, significant speech delay was reported in most individuals (n ¼ 29)
even in those with IQ in the borderline range. The finding that verbal IQ was
reduced relative to performance IQ in three of the five individuals for whom
deep neuropsychological phenotyping was available adds further evidence to our
observation that expressive language is particularly affected in this disorder
How were the 34
individuals identified?
In the Table 1 of the paper, we see
that the great majority of the children had been identified from WES (whole
exome sequencing), a few had WGS (whole genome sequencing) and just one via
micro array testing.
They families clearly opted to share
their data, in the hope of some researcher finding it useful later, as Chopra, Amiel and
Gordon clearly did after a few years later.
How do you figure
out the DEGs (differentially expressed genes)?
To treat ANKRD17 deficiency (now known as Chopra Amiel Gordon Syndrome) you have a choice.
·
Increase expression of ANKRD17 via gene therapy, or a drug (if that
were possible)
·
Treat some of the
downstream DEGs (Differentially Expressed Genes)
Mary asked how you could identify the
DEGs, given there is only one paper published on Chopra Amiel Gordon Syndrome.
You can start by reviewing everything known about ANKRD17.
A very good place to start is on the
GeneCards website.
https://www.genecards.org/cgi-bin/carddisp.pl?gene=ANKRD17
Most people will
end up having to learn some new words to understand everything on the above website.
The first thing to note is just how
wide ranging are the functions of this gene and this accounts from the wide-ranging
problems associated with it. It even plays
a role in dealing with both viral and bacterial infections.
It is particularly upregulated in the
fetal brain and that likely leads to the autism/ID related effects.
Protein
differential expression in normal tissues from HIPED for ANKRD17 Gene
This gene is overexpressed in Lung (18.9), Platelet (15.4),
Retina (8.4), and Fetal Brain (6.4).
We can see that this gene is
associated with Chopra Amiel Gordon Syndrome and
Non-Specific Syndromic Intellectual Disability.
Quite possibly, Non-Specific Syndromic Intellectual
Disability was used as a term because Chopra Amiel Gordon Syndrome did
not yet exist.
But is useful to look up Non-Specific Syndromic Intellectual
Disability, to see which other genes are listed. This then tells you much about what can cause
ID. Follow the link below.
https://www.malacards.org/card/non_specific_syndromic_intellectual_disability
We see a very long list of syndromes and genes.
There are 61 genes listed.
Going back to the Genecards ANKRD17 page, we can see
if there are known protein interactions that might result in autism/ID.

EIF4E2 does look familiar, and I recall a link to Fragile X. So, I looked it up.
Note that we see both EIF4E and EIF4E2 - Eukaryotic Translation Initiation Factor 4E Family Member 2. Note that is has a second name, 4EHP.
EIF4E2 is a version/homolog of EIF4E
EIF4E2 = 4EHP
The eIF4E homolog
4EHP (eIF4E2) regulates hippocampal long-term depression and impacts social behavior
Background: The regulation of protein synthesis is a critical
step in gene expression, and its dysfunction is implicated in autism spectrum
disorder (ASD). The eIF4E homologous protein (4EHP, also termed eIF4E2) binds
to the mRNA 5' cap to repress translation. The stability of 4EHP is maintained through
physical interaction with GRB10 interacting GYF protein 2 (GIGYF2).
Gene-disruptive mutations in GIGYF2 are linked to ASD, but causality is
lacking. We hypothesized
that GIGYF2 mutations cause ASD by disrupting 4EHP function.
4EHP is
expressed in excitatory neurons and synaptosomes, and its amount increases
during development. 4EHP-eKO mice display exaggerated mGluR-LTD, a phenotype
frequently observed in mouse models of ASD.
Conclusions: Together these
results demonstrate an important role of 4EHP in regulating hippocampal
plasticity and ASD-associated social behaviors, consistent with the link
between mutations in GIGYF2 and ASD.
The disruption of protein
synthesis (mRNA translation or translation) in the brain by genetic
perturbations of its regulators constitutes a known underlying etiology for ASD
[2, 3]. For
most mRNAs, initiation of translation requires binding of initiation factors to
their 5′ end at a modified guanine nucleotide (m7GpppN, where N is
any nucleotide) termed the 5′ cap [4]. The
eukaryotic initiation factor (eIF) 4F complex is comprised of the cap binding
protein eIF4E, an mRNA helicase eIF4A, and a molecular scaffold eIF4G. Together
these proteins facilitate recruitment of the ribosomal 43S preinitiation
complex to the mRNA. Overactivity
of eIF4E in humans has been implicated in ASD [5, 6] and ASD-like phenotypes in mice [7, 8]. Indeed, disruption of the proteins regulating eIF4E
activity, such as fragile
X mental retardation protein (FMRP)
[9], cytoplasmic FMR1 interacting protein 1 (CYFIP1) [10], and eIF4E-binding protein 2 (4E-BP2) [8, 11, 12], is implicated in ASD. It is therefore necessary to
investigate the function of ASD-linked genes that encode for regulators of translation.
Whole-genome sequencing of ASD patients has been invaluable in identifying
these genes.
If you look up the protein interaction for the Fragile X gene (FMRI), you do indeed see EIF4E close by. FMR1 encodes the fragile X mental retardation protein.
This blog is full of ideas regarding treating Fragile X,
because there are so many studies of that type of autism.
It is rather mind-boggling that there are still no approved
therapies for Fragile X. The same holds
true for Down Syndrome (DS). This is a
recurring story, where it pays to be the early adopter, not one of the passive
followers.
Leaky
ATP from either Mitochondria or Neurons in Fragile X and Autism
In that post I suggested “Mirapex - a
miracle for Fragile X?”
In the post below we saw how EIF4E leads to autism, and how
FMRP from Fragile-X affects EIF4E.
Vasopressin,
Oxytocin, the Lateral Septum, Aggression and Social Bonding, Autism gene NLGN3
and MNK inhibitors for reversing Fragile-X and likely more Autism

One of the papers below goes further
and suggests
“This
work uncovers an unexpected convergence between the genetic autism risk factor
Nlgn3, translational regulation, oxytocinergic signalling, and social novelty
responses”
“We
propose that pharmacological inhibition of MNKs may provide a new therapeutic
strategy for neurodevelopmental conditions with altered translation
homeostasis”
“Our
work not only highlights a new class of highly-specific, brain-penetrant MNK
inhibitors but also expands their application from fragile X syndrome to a
non-syndromic model of ASD”
Regarding
Fragile X
“Collectively, this work establishes eFT508 (an MNK inhibitor) as a
potential means to reverse deficits associated with FXS.”
Conclusion
My quick
look at the subject suggests that, amongst other likely DEGs, the NLGN (neuroligin)
genes are quite possibly miss-expressed.
In
humans, alterations in genes encoding neuroligins are implicated in autism and other cognitive disorders.
In
Genecards the association is with EIF4E2 rather than the EIF4E, which we
know affects neuroligin expression. But EIF4E2 is just a version of EIF4E.
These protein interaction maps are not perfect and different
sources often come up with slightly different maps.
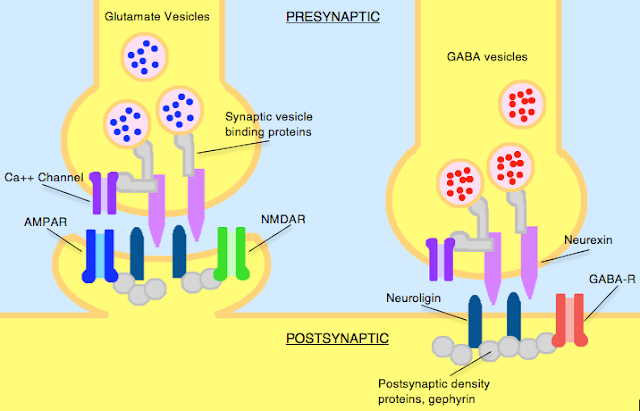
What are Neurexins and Neuroligins?
Neurexins and neuroligins are synaptic cell-adhesion molecules that connect pre- and postsynaptic neurons at synapses, they mediate signalling across the synapse, and shape the properties of neural networks by specifying synaptic functions. Neurexins and neuroligins are therefore very important and can be dysfunctional in autism.
It looks
like growth signaling is disturbed in Chopra-Amiel-Gordon Syndrome, but it not always in the same way. Both too much and too
little growth are possible.
An MRI
would not be a bad idea, and measuring the corpus callosum would be helpful.
The corpus callosum connects the right and left side of the brain and is the largest
white matter structure in the brain, which means lots of myelin should be
there.
If it is very narrow, that would tell you something, hopefully it is normal. You cannot really change its size, but if it lacked myelination that might be something you could affect.
Positive Correlations
between Corpus Callosum Thickness and Intelligence
Trying the cheap and partially effective treatments for fragile X might be helpful. It is possible that the Fragile X DEGs overlap with the Chopra-Amiel-Gordon Syndrome DEGs.
The
following drugs are cheap generics that are helpful, to some extent, in
Fragile-X.
·
Metformin
·
Lovastatin
·
Baclofen
As the altered E/I balance is present in Fragile X and most autism, it would be worthwhile trying the E/I corrective therapies that exist, in case one is beneficial. There are different causes of an E/I imbalance, but since there are not many therapies, it is easier to just try them one by one.
It is
also highly likely that common features of autism may be present, such as
·
oxidative stress (NAC)
·
neuroinflammation (numerous therapies)
·
impaired myelination (Clemastine, Ibudilast, NAG) NAG
is not the same as NAC, it is N-acetylglucosamine
·
mitochondrial dysfunction (Carnitine, antioxidants, activate
PGC-1 alpha via PPAR gamma e.g. with Pioglitazone)
·
folate receptor antibodies (Calcium folinate)
If the
Corpus Callosum is smaller than it should be, or is demyelinated, you could try
high bio-availability curcumin, in addition to the above pro-myelination
therapies.
Which ion
channel dysfunctions appear in Chopra-Amiel-Gordon Syndrome? I did not see any
clues, but where there is epilepsy, there is very likely going to be an ion
channel dysfunction involved.