
Today’s post takes another dip into the genetics of autism and currently existing therapies that could be re-purposed for autism. We also see that many secrets remain beyond the 3% of your DNA that usually gets all the research attention. The remaining 97% is not junk after all.
There was an earlier post on this blog that introduced Epigenetics. It is not such a complicated subject, just think about it as little tags on your DNA that turn genes on/off usually when they should not be, but there remains the possibility to use epigenetics for good. In people with under-expression of an important gene you could “tag it” and then increase its expression.
The exome is the part of your DNA that encodes the various proteins needed to build your body. The remaining 97% of your DNA was once thought to be just junk; we saw in recent post that one part contains enhancers and silencers that control expression of the genes in the 3% that is the exome.
A recent study of gene expression in neurological conditions including autism showed just how broadly disturbed gene expression is.

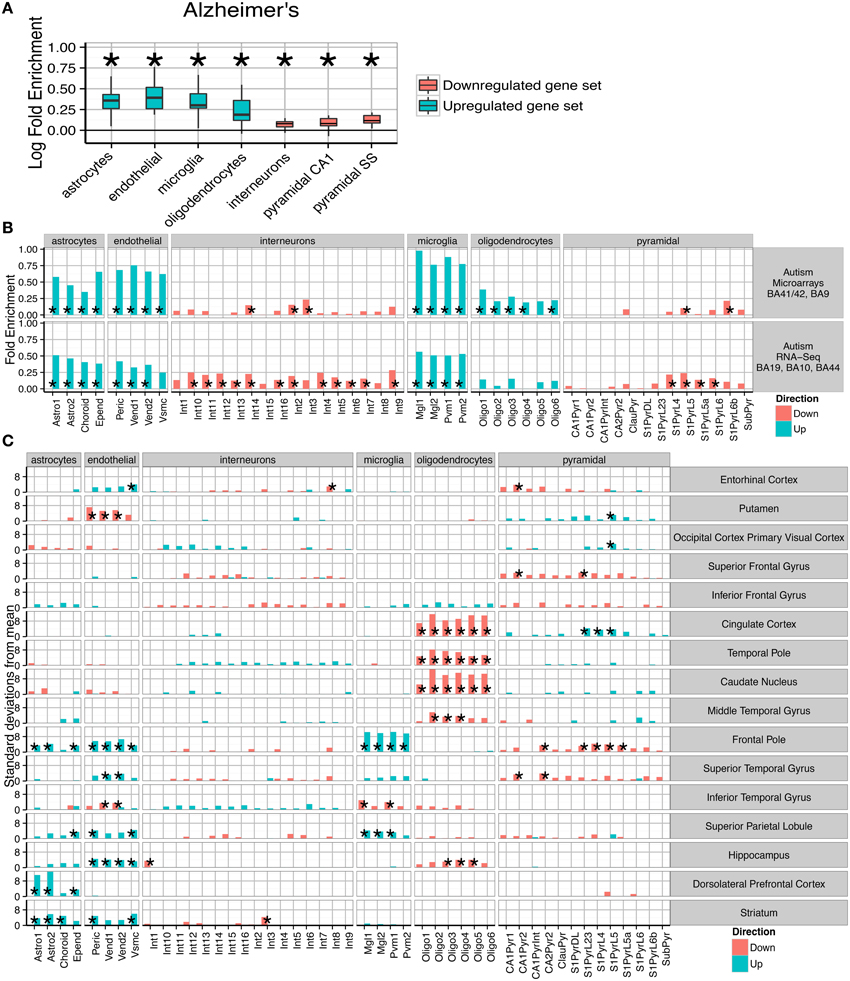
(A) Consistent fold enrichments were found for each cell type across fourteen cortical and three subcortical brain regions of Alzheimer's patients. The box plots mark the distribution of cellular fold enrichments across all the brain regions examined. Asterisks mark that the fold enrichment for each cell type that was found to be significantly non-zero with p < 0.05. (B) Two independent autism studies show the same cellular phenotypes, including upregulation of glial cells and downregulation of neurons. Asterisks mark those cell types found to be significantly differential with p < 0.05 after BH correction over all groups.
Here I am making the point that even though only a handful of genes may have an identifiable dysfunction, a much broader range of genes seem to be affected, as we see in the wide range of over and under expressed genes.
While it would be logical to think about a specific dysfunction needing a therapy that targets just that gene, this appears not to be necessary.
It appears that downstream processes may be the most damaging/relevant, for example disturbances in Protein Kinase A and C (PKA and PKC) may play a key role in many cases of regressive autism, and this will feature in its own post, because it would be treatable today.
Reduced activity of protein kinase C in the frontal cortex of subjects with regressive autism: relationship with developmental abnormalities.
Brain Region–Specific Decrease in the Activity and Expression of Protein Kinase A inthe Frontal Cortex of Regressive Autism
Both the above papers are by Abha and Ved Chauhan. I put Abha on my Dean’s list long ago. I did have a discussion with her a while back. She is clearly a very nice person and intellectually towers over the Curemark lady (Joan Fallon) who gets $40 million to play with her pancreatic enzymes, but never publishes anything except very superficial patents.
I think for $40 million Abha and Ved could figure it all out.
PKB, otherwise known as Akt is also very relevant to some types of autism.
Tamoxifen, recently shown to reverse autism in a SHANK3 mouse model, is a PKC inhibitor.
Another epigenetic drug, Theophylline activates PKA.
Akt, also known as protein kinase B (PKB), is a central node in cell signaling downstream of growth factors, cytokines, and other cellular stimuli. Aberrant loss or gain of Akt activation underlies the pathophysiological properties of a variety of complex diseases, including type-2 diabetes and cancer.
If you could identify if a particular person was hypo/hyper in PKA, PKB and PKC, this might well open the door to an effective treatment.
Research on PKB, also known as AKT
Dysregulation of theIGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders.
And a paper from the clever Japanese:-
Autism spectrum disorder is a set of neurodevelopmental disorders in terms of prevalence, morbidity and impact to the society, which is characterized by intricate behavioral phenotype and deficits in both social and cognitive functions. The molecular pathogenesis of autism spectrum disorder has not been well understood, however, it seems that PI3K, AKT, and its downstream molecules have crucial roles in the molecular pathogenesis of autism spectrum disorder. The PI3K/AKT signaling pathway plays an important role in the regulation of cell proliferation, differentiation, motility, and protein synthesis. Deregulated PI3K/AKT signaling has also been shown to be associated with the autism spectrum disorder. Discovery of molecular biochemical phenotypes would represent a breakthrough in autism research. This study has provided new insight on the mechanism of the disorder and would open up future opportunity for contributions to understand the pathophysiology
For those who favour dietary intervention:-

Based on the above chart curcumin should likely be good for my N=1 case of autism. Time will tell.
Consequences of upstream dysfunctions
So it might be better to consider autism as a disease of wider downstream gene expression, rather than necessarily of “faulty” genes. Modulating the resulting wider gene expression may be much more realistic than fixing individual genes.
It is certainly plausible that the body has its own protective self-repair mechanism that might be somehow re-energized. Some people have pondered why so many highly intelligent mathematicians and computer scientists seem have relatives with autism. The clever genes do associate with a type of autism plus ID/MR. It was suggested that protective genetic changes might be in play, so that the people with the most genetic variance are actually the family members without the autism.
This does remain conjecture, but as more whole genome data is collected we are seeing some interesting findings.
A fascinating very recent study that looked at a group of 53 families with autism using the traditional approach of whole exome sequencing and also microarray.
Using these methods, that are the current gold standard, the researchers found very little. Dysfunctions in the 700 known autism genes were not detected.
However using more expensive whole genome sequencing, dysfunctions were identified in the “DNA junk” zone very close beside the known autism genes. The researchers were then able to identify the genetic cause of 30% of the cases, a big improvement on 0%. I expect if they looked a little harder the 30% would be higher.
“We performed whole-genome sequencing (WGS) of 208 genomes from 53 families affected by simplex autism.”
“For the majority of these families, no copy-number variant (CNV) or candidate de novo gene-disruptive single-nucleotide variant (SNV) had been detected by microarray or whole-exome sequencing (WES).
Comparing the sequences of the individuals with autism and those of their unaffected siblings, the researchers found that people with autism are more likely to have genetic variants — either single base-pair changes in the sequence or small CNVs — in swaths of DNA abutting known autism genes. But the researchers only found the variants after they restricted their search to regions of the genome already implicated in autism, and even then the statistical significance is modest.
Sequencing whole genomes could reveal the genetic cause of autism in as much as 30 percent of people for whom faster and cheaper sequencing methods come up short
“It’s increasing power even in areas that are supposed to be covered by whole-exome sequencing,” says Peixoto. “It seems that it’s clear that whole-genome sequencing will become the standard.”
Epigenopathies
Many diseases have an epigenetic component. The severe progressive asthma that is COPD is a well-known example. It appears that smoking in middle age often leads to permanent epigenetic changes that come back to haunt often then non-smokers in old age. Even though they have not smoked for twenty years, there oxidative stress response has been permanently modified. This results in a kind of steroid resistance, so that usually reliable drug therapies fail to work.
It is thought that autism has an epigenetic component. This would do some way to explaining 30-40% of the increase in prevalence in recent years that is not explained by ever widening diagnostic criteria.
Because epigenetic changes can be heritable and can be accumulated from all kinds of exposures, even simple ones like severe emotional stress and pollution, you can reconcile autism as being primarily a genetic condition even though incidence has clearly risen within one or two generations. So you can have an “epigenetic epidemic”, so to speak.
Epigenetics as a therapy
While much is written about epigenetic change being bad, it could also be good.
There are many known substances that affect gene expression; some are very target specific which is useful.
This answers a recent issue raised by a reader of this blog who did exome sequencing. What is the point of discovering a genetic dysfunction if there is no therapy? Medicine is some decades behind science, better to know what gene is affected because you well be able to affect its expression, you just need some help from Google.
This answers a recent issue raised by a reader of this blog who did exome sequencing. What is the point of discovering a genetic dysfunction if there is no therapy? Medicine is some decades behind science, better to know what gene is affected because you well be able to affect its expression, you just need some help from Google.
Epigenetic therapy could be used to remove unwanted tags, but it could also be used to leave new ones to upregulate under-expressed genes.
Such epigenetic therapy is already a reality in COPD and is being considered for rare single autisms where one copy of the gene is not functional, so turn up the volume on the remaining copy.
As we saw in the post on epigenetics, one potential category of drugs are HDAC inhibitors, these would affect one epigenetic mechanism.
There are many such HDAC inhibitors and most have other modes of action, so you cannot be sure what is giving the noted effect.
Valproate
This epilepsy drug has numerous effects including as a HDAC inhibitor. Given to mothers during pregnancy it can cause autism in the offspring, but when given to the affected offspring the autism can be reduced.
Valproate is given off label to treat autism even when no epilepsy is present.
As we saw in the comments section, long term valproate se can have side effects.
Sulforaphane
This substance derived from broccoli and patented by Johns Hopkins, is another HDAC inhibitor. It also upregulates Nrf2, which turns on the oxidative response genes. This was proposed as a COPD therapy by Professor Barnes.
We saw in a post that for Nrf2 to have its full effect there needed to be enough of a protein called DJ-1. You can increase DJ-1 expression with cinnamon (sodium benzoate).
That was one reason to think that cinnamon would complement Sulforaphane as a therapy for both COPD and some autism.
Sodium Butyrate
Sodium Butyrate is an HDAC inhibitor that is available as a supplement. We came across it in an earlier post as a precursor to butyric acid. Butyric acid plays a role in the permeability of the gut and the Blood Brain Barrier (BBB). It also seems to protect from auto immune disease.
Butyrate is fed to millions of farm animals every day to increase their resistance to auto-immune disease.
Butyric acid is produced naturally in the gut by the bacteria living there, however the amount can be increased by the uses of a particular probiotic-bacteria.
This would support the uses of sodium butyrate and the Miyari 588 bacteria.
I have on my to-do-list to investigate higher doses of Miyari 588, but having read the comment by Alli that 500 mg of sodium butyrate is effective, I will try that first. She also found higher doses ineffective, which was the same in a mouse study published last November,
The study below highlights which genes were down-regulated and which were up-regulated, the overall effect was beneficial
Sodium butyrate attenuate ssocial behavior deficits and modifies the transcription ofinhibitory/excitatory genes in the frontal cortex of an autism model.
The core behavioral symptoms of Autism Spectrum Disorders (ASD) include dysregulation of social communication and the presence of repetitive behaviors. However, there is no pharmacological agent that is currently used to target these core symptoms. Epigenetic dysregulation has been implicated in the etiology of ASD, and may present a pharmacological target. The effect of sodium butyrate, a histone deacetylase inhibitor, on social behavior and repetitive behavior, and the frontal cortex transcriptome, was examined in the BTBR autism mouse model. A 100 mg/kg dose, but not a 1200 mg/kg dose, of sodium butyrate attenuated social deficits in the BTBR mouse model. In addition, both doses decreased marble burying, an indication of repetitive behavior, but had no significant effect on self-grooming. Using RNA-seq, we determined that the 100 mg/kg dose of sodium butyrate induced changes in many behavior-related genes in the prefrontal cortex, and particularly affected genes involved in neuronal excitation or inhibition. The decrease in several excitatory neurotransmitter and neuronal activation marker genes, including cFos Grin2b, and Adra1, together with the increase in inhibitory neurotransmitter genes Drd2 and Gabrg1, suggests that sodium butyrate promotes the transcription of inhibitory pathway transcripts. Finally, DMCM, a GABA reverse agonist, decreased social behaviors in sodium butyrate treated BTBR mice, suggesting that sodium butyrate increases social behaviors through modulation of the excitatory/inhibitory balance. Therefore, transcriptional modulation by sodium butyrate may have beneficial effects on autism related behaviors.
Theophylline
Theophylline is an old asthma drug that is an HDAC inhibitor.
At low doses it is now being trialled as an epigenetic add-on therapy in COPD. It pretty obviously does work, but data needs to be collected to measure how effective it is and what is the best dose.
It shows how the COPD researchers/clinicians like Professor Barnes are doing a good job and not frightened to experiment.
Would a similar low dose of theophylline benefit a sub-group of those with autism/schizophrenia? I think it is quite likely.
COPD and autism/schizophrenia share the same impaired oxidative stress response.
Chronic Obstructive Pulmonary Disease (COPD) is a progressive lung disease characterised by progressive airflow limitation. In the UK, it affects around 3 million people, is the fifth leading cause of death and costs the NHS approximately £1 billion annually. Exacerbations of COPD account for 60% of NHS COPD costs and are associated with accelerated rate of lung function decline, reduced physical activity, reduced quality of life, increased mortality and increased risk of co-morbidities. COPD treatment guidelines recommend inhaled corticosteroids (ICS) to reduce exacerbations and improve lung function. However, in COPD, airway inflammation is relatively insensitive to the anti-inflammatory effects of ICS and even high doses fail to prevent exacerbations. Preclinical and pilot studies demonstrate that low dose theophylline may increase the sensitivity of the airway inflammation to ICS, and thus when used with ICS will reduce the rate of COPD exacerbation. In this study we will determine the clinical effectiveness and cost-effectiveness of adding low dose theophylline to ICS therapy in patients with COPD. The primary outcome is the number of exacerbations. The primary economic outcome is the cost-per-QALY gained during the one year treatment period. We will recruit 1424 participants from primary and secondary care across seven areas of the UK. Participants will be randomised to theophylline (200 mg once or twice daily depending on smoking status and weight) or placebo for 12 months. We will follow participants up at six and twelve months to assess the number of exacerbations. We will also collect data on adverse events, health care utilisation, quality of life and breathlessness, and lung function. Low dose theophylline is cheap (10p/day) and, if shown to make current ICS therapy more effective in a cost effective manner, it will improve the quality of life of COPD patients and reduce the burden of COPD on the NHS.
At large doses, Theophylline has long been a therapy for asthma and COPD, but as with Sodium Butyrate, it is quite possible that larger doses of Theophylline produce a different result. In other words the epigenetic effect fortunately comes from the low dose.
Low doses mean less chance of side effects.
For example, in anyone predisposed to reflux/GERD/GORD many asthma drugs pose a problem because at the same time as opening the airways in your lungs they will relax the lower esophageal sphincter and allow stomach acid to rise upwards.
We saw in an earlier post that in some types of autism something called mGluR5 is dysfunctional in the brain. By chance mGluR5 is also involved in closing the lower esophageal sphincter. In people with reflux/GERD/GORD a mGluR5 inhibitor was found to have promise for the management of their symptoms.
Randomised clinical trial:effects of monotherapy with ADX10059, a mGluR5 inhibitor, on symptoms and reflux events in patients with gastro-oesophageal reflux disease.
So it is not surprising that many people with autism also have reflux/GERD/GORD.
But the dysfunction with mGluR5 in autism can be both hyper and hypo, so the therapy might be a positive allosteric modulator (PAM), or a negative allosteric modulator (NAM).
In someone with autism + reflux/GERD/GORD it would be reasonable to think a NAM, like ADX10059, might help both conditions.
But the dysfunction with mGluR5 in autism can be both hyper and hypo, so the therapy might be a positive allosteric modulator (PAM), or a negative allosteric modulator (NAM).
In someone with autism + reflux/GERD/GORD it would be reasonable to think a NAM, like ADX10059, might help both conditions.
Gene Repression and Genome Stability
There is another epigenetic process that may be disturbing gene expression in some people and may be treatable.
I have been trying to find why so many people with autism can benefit from biotin; I think I have found a plausible explanation.
“Biotinylation of histones plays a role in gene repression and repression of transposable elements, thereby maintaining genome stability”
I think in some people with autism and no clinical deficiency of biotin the continued “overdosing” of biotin might be having an effect on gene expression, bringing things a little closer to where they should be.
Rather beyond the scope of this blog, it appears that in some people the impaired genome stability, reversible with biotin(ylation), this might be a significant cancer risk.
In essence, for most people supraphysiological concentrations of biotin will do absolutely nothing, but in a sub-group it might do a lot of good. It is epigenetic, but you do not have to understand it to benefit from it. It is complicated.
Transposable elements such as long terminal repeats (LTR) constitute ∼45% of the human genome; transposition events impair genome stability. Fifty-four promoter-active retrotransposons have been identified in humans. Epigenetic mechanisms are important for transcriptional repression of retrotransposons, preventing transposition events, and abnormal regulation of genes. Here, we demonstrate that the covalent binding of the vitamin biotin to lysine-12 in histone H4 (H4K12bio) and lysine-9 in histone H2A (H2AK9bio), mediated by holocarboxylase synthetase (HCS), is an epigenetic mechanism to repress retrotransposon transcription in human and mouse cell lines and in primary cells from a human supplementation study. Abundance of H4K12bio and H2AK9bio at intact retrotransposons and a solitary LTR depended on biotin supply and HCS activity and was inversely linked with the abundance of LTR transcripts. Knockdown of HCS in Drosophila melanogaster enhances retrotransposition in the germline. Importantly, we demonstrated that depletion of H4K12bio and H2AK9bio in biotin-deficient cells correlates with increased production of viral particles and transposition events and ultimately decreases chromosomal stability. Collectively, this study reveals a novel diet-dependent epigenetic mechanism that could affect cancer risk.
Here, we provide evidence for the existence of a novel diet-dependent epigenetic mechanism that represses retrotransposons. Importantly, we demonstrated that depletion of biotinylated histones in biotin-deficient cells increases LTR transcript levels, production of viral particles, and retrotransposition events, and ultimately decreases chromosomal stability. Both biotin deficiency and supplementation are prevalent in the US. For example, moderate biotin deficiency has been observed in up to 50% of pregnant women (35,36). About 20% of the US population reports taking biotin supplements (37), producing supraphysiological concentrations of vitamin in tissues and body fluids (23,28,35). The findings presented here suggest that altered biotin status in these population subgroups might affect chromosomal stability and cancer risk.
Biotin and biotinidase deficiency
Biotin requirements for DNA damage prevention
Conclusion
I never got round to writing part 2 of my epigenetics post, but my experience of HDAC inhibitors to date has been very positive.
I would be the first to admit that this is rather hit and miss. It was only when reading the paper on potential therapies for Pitt Hopkins, that was openly musing about HDAC inhibitors, in an equally hit and miss approach, that I thought I would write further about it.
It really seems totally haphazard, because you cannot predict the effect with any level of certainty. If there is a self-repair mechanism trying to maintain homeostasis of the genome, haphazard may be good enough.
10mg of biotin twice a day does have a mild but noticeable stabilizing effect; is this caused by better maintaining genome stability? I have no idea.
I will try sodium butyrate and if it works I will have to establish what dose of Miyari 588 produces the same effect. Both are used in animal feed to reduce inflammatory disease, so you are already indirectly exposed to them if you eat meat.
Theophylline should also be investigated. This is a very well understood drug and small doses really do seem to help people with COPD.
PKA, PKB and PKC are likely at the core of most people’s autism. Many existing therapies can modify their expression.
Whole genome sequencing, carried out at great precision, is clearly the only satisfactory genetic testing method. The other, cheaper, methods are just missing key data and giving many false negative results, i.e. saying there are no identifiable genetic dysfunctions, when this is not true.


{kind=link}