“three cytokine levels, namely the IFN-γ,
MIG and IFN-α2 … These cytokine
levels at the baseline could improve the prediction of the bumetanide
responders”
“… cytokines had a potential to construct a
blood signature for predicting and monitoring the bumetanide treatment in young
children with ASD.”
“a significant part of the clinical
heterogeneity in the treatment effect of bumetanide for ASD is associated with
the differences in the immune system of patients”
Autism is a
very heterogeneous family of conditions and this is a big part of the reason
why all clinical trials to date have failed.
Ideally, there would be a diagnostic test to identify which person will
respond to which therapy. Then you can
have a successful clinical trial, because you are only including people likely
to respond.
Researchers
from China have just published their results that suggest that a blood test
measuring three inflammatory markers can predict who will respond to
bumetanide. This is good news and where
it is coming from is also very notable.
Autism
research has been very fragmented, some of it is very sophisticated and
insightful but much is very amateur and some is quite trivial. There is usually a real lack of common sense
among these people and no sense of urgency whatsoever.
China is a
very organised country; plans are made and then they are implemented. Forget political correctness.
This kind of
approach is what is required to move along with autism treatment.
In addition,
there is also another new study from China, this time on the microbiota in
autism that compared those with and without GI problems (it found it is equally
disturbed in both groups). Hopefully, that Chinese group will do the next
common sense step and compare the microbiota of autistic people with and
without restrictive diets. To what extent to people give themselves a
microbiota problem through poor diet.
Disentangling the relationship of gut microbiota,
functional gastrointestinal disorders and autism: a case–control study on
prepubertal Chinese boys
The altered gastrointestinal microbiota composition in ASD appeared to be independent of comorbid functional gastrointestinal disorder
The
bumetanide researchers are from Fudan
University in Shanghai, one of the 3 ultra-selective Chinese Universities
alongside Tsinghua University and Peking University in Beijing.
The paper, not surprisingly, may look complicated,
but there are a great deal of interesting things in it.
In their words:-
An immuno-behavioural covariation was
identified between symptom improvements in the Childhood Autism Rating Scale
(CARS) and the cytokine changes among interferon (IFN)-γ, monokine
induced by gamma interferon and IFN-α2. Using this covariation, three
groups with distinct response patterns to bumetanide were detected
The three groups were: best
responders, least responders and medium responders.
It should be noted that the dosage
used in their trials was 0.5mg of bumetanide twice a day.
Chinese children tend to be smaller
than Western children and this might help explain why the results were more
positive than in Servier’s failed phase 3 clinical trial in Europe. I also
imagine the Chinese children were more severely autistic than the European
group.
The dosage used is selected to
minimize the diuresis rather than to maximize the impact on the autism. This is
understandable, but I think it is a mistake.
Bumetanide, a drug being studied in autism
spectrum disorder (ASD) may act to restore gamma-aminobutyric acid (GABA) function,
which may be modulated by the immune system. However, the interaction between
bumetanide and the immune system remains unclear. Seventy-nine children with
ASD were analysed from a longitudinal sample for a 3-month treatment of
bumetanide. The covariation between symptom improvements and cytokine changes
was calculated and validated by sparse canonical correlation analysis. Response
patterns to bumetanide were revealed by clustering analysis. Five classifiers
were used to test whether including the baseline information of cytokines could
improve the prediction of the response patterns using an independent test
sample. An
immuno-behavioural covariation was identified between symptom improvements in
the Childhood Autism Rating Scale (CARS) and the cytokine changes among
interferon (IFN)-γ, monokine induced by gamma interferon and IFN-α2.
Using this covariation, three groups with distinct response patterns to
bumetanide were detected, including the best (21.5%, n = 17;
Hedge’s g of improvement in CARS = 2.16), the least (22.8%, n = 18; g = 1.02)
and the medium (55.7%, n = 44; g = 1.42) responding
groups. Including the cytokine levels significantly improved the prediction of
the best responding group before treatment (the best area under the curve,
AUC = 0.832) compared with the model without the cytokine levels (95%
confidence interval of the improvement in AUC was [0.287, 0.319]). Cytokine measurements can help
in identifying possible responders to bumetanide in ASD children, suggesting
that immune responses may interact with the mechanism of action of bumetanide
to enhance the GABA function in ASD.
The use of bumetanide as a potential drug to improve
symptoms in ASD is based on a hypothesised pathoetiology of ASD, namely the
delayed developmental switch of the gamma-aminobutyric acid (GABA) functioning
from excitatory to inhibitory [10,11,12]. In the valproate and fragile X rodent models of
autism, this GABA-switch can be facilitated by the reduction of intracellular
chloride concentration, which is mediated by a sequential expression of the
main chloride transporters, such as the potassium (K)-Cl co-transporters 2
(KCC2) and the importer Na-K-Cl cotransporter 1 (NKCC1) [12]. Therefore, bumetanide as an NKCC1 inhibitor has
been tested for its ability to restore GABA function in ASD [5,6,7, 13, 14]. However, these transporters can also be influenced by other molecules,
such as cytokines, which are a number of small cell-signalling proteins closely
interacting with each other to modulate the immune reactions. The
cytokines have been implicated not only in brain development [15], but also in GABAergic transmission [16,17,18]. It has been reported that the interferon (IFN)-γ can
decrease the levels of NKCC1 and the α-subunit of Na+-K+-ATPase,
contributing to the restore of inhibitory GABA function [16]. In mice subjected to maternal deprivation, the
interleukin (IL)-1 has also been found to reduce the expression of KCC2,
delaying the developmental switch of the GABA function and thereby possibly
contributing to the pathophysiology of developmental disorders such as ASD [17, 18]. Therefore, a question naturally arises that
whether the treatment effect of bumetanide for ASD can be affected by the
immune responses in the patients.
Indeed, compared with healthy controls, changes of
the cytokine levels have already been reported in patients with ASD [19,20,21,22]. Recent meta-analyses showed that the levels of
anti-inflammatory cytokines IL-10 and IL-1 receptor antagonist (Ra) were
decreased [20], while proinflammatory cytokines IL-1β,
IL-6 and anti-inflammatory cytokines IL-4, IL-13 were elevated in blood of
patients with ASD [21]. The levels of IFN-γ, IL-6, tumour necrosis
factor (TNF)-α, granulocyte-macrophage colony-stimulating factor
(GM-CSF) and IL-8 were observed to be elevated [22] in postmortem brain tissues of ASD patients, and
increased level of IFN-γ, monocyte chemotactic protein (MCP)-1, IL-8,
leukaemia inhibitory factor (LIF) and interferon-gamma inducible protein
(IP)-10 were found in another study [23]. These widely spread changes suggest that the
cytokine signalling in ASD may be better characterised by multivariate patterns
of cytokines. In literatures, many associations had been reported between the
levels of cytokines (e.g., MCP-1, IL-1β, IL-4, IL-6, etc.) and both core
symptoms and adaptive functions in children with ASD [24,25,26]. Therefore, it has been suggested that cytokines may be used as
biomarkers to identify different subsets within ASD. In each of these subsets
the patients with ASD may share a commonly immune-related pathoetiology and
therefore may have similar profiles of response to treatment [27].
Based on
these previous findings, we analysed data acquired through the Shanghai Xinhua
ASD registry, China, that began in 2016 to test the hypothesis that the immune
activity of patients might help to identify the best responders to bumetanide
in ASD.
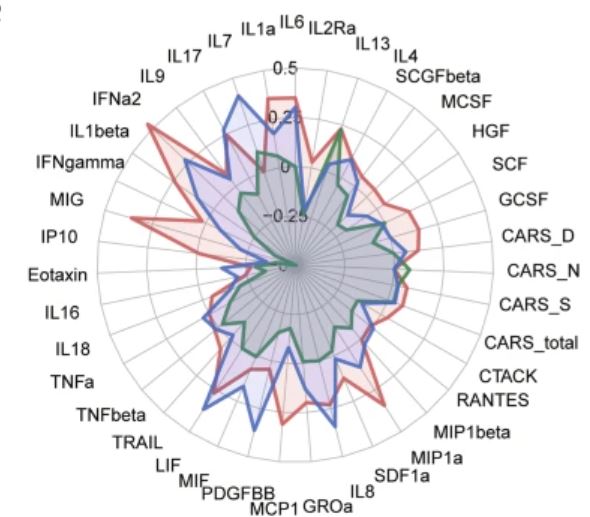
Between May 1st, 2018, to April 30th, 2019, a total of 90 ASD children, aged 3–10 years old, under a 3-month stable treatment of bumetanide without behavioural interventions and any concomitant psychoactive medications had both blood draws and behavioural assessments. Among these patients, 11 of them were further excluded due to the lack of the follow-up data at month 3. A group of 37 children, under 3-month stable treatment of placebo without behavioural interventions and any concomitant psychoactive medications had both blood draws and behavioural assessments. Therefore, the current analysis used a subsample of 116 young children with ASD, whose blood samples were available both before and after the treatment. The blood samples were sent in three batches (Discovery Set: n = 37 on December 4, 2019; Validation Set: n = 42 on May 22, 2019; and Control Set: n = 37 on January 5, 2022) to measure the serum levels of 48 cytokines for the immune response (Table S1), and the clinical symptoms were assessed using CARS, ADOS and the Social Responsiveness Scale (SRS).
In this study, we observed a
significant improvement of clinical symptoms with bumetanide treatment in
children with ASD, and such improvement was associated with a pattern of
changes in three cytokine levels, namely the IFN-γ, MIG and IFN-α2
(r = 0.459 in the Discovery Set and r = 0.316 in the
Validation Set). These cytokine levels at the baseline could improve the
prediction of the bumetanide responders compared with using the behavioural
assessments alone, and the best predictor achieved an AUC of 0.83 in the
independent test data set (Table S8).
The implications of these findings may be twofold: (1) a significant part of
the clinical heterogeneity in the treatment effect of bumetanide for ASD is
associated with the differences in the immune system of patients, and (2) the
component score of cytokines had a potential to construct a blood signature for
predicting and monitoring the bumetanide treatment in young children with ASD.
Following the protocols of previous studies [8], bumetanide treatment consisted of two 0.5 mg tablets
per day for three months, given at 8:00 a.m. and 4:00 p.m. The tablet size is
8 mm diameter x 2 mm thickness, which is quite small. Each time, the patient
took half of a tablet, which was not difficult for most of the patients.
However, the careers were recommended to grind the half-tablet into powder and
give the powder in water, if necessary. Possible side effects were closely
monitored during the treatment. Blood parameters (serum potassium and uric
acid) were monitored via laboratory tests (Table S2) and symptoms (thirst, diuresis, nausea, vomiting,
diarrhoea, constipation, rash, palpitation, headache, dizziness, shortness of
breath, and any other self-reported symptoms) were telephone interviewed
(Table S3), and both of them were reported to the research
team by telephone at 1 week and 1 month after the initiation of treatment and
at the end of the treatment period. The cytokine levels of the children with gastrointestinal problems were
compared with those without such problems (Table S4).
The supplemental table S4 shows that GI
problems had no effect on cytokine levels.
Changes
after the administration of bumetanide
Seventy-nine patients were treated with bumetanide
for 3 months, and the CARS total score decreased after the treatment (effect
size Cohen’s d = 1.26, t78 = 11.21, p < 0.001).
The treatment effect showed no difference between the Discovery Set and the
Validation Set (ΔCARS_total: mean(±SD): 1.54 (±1.40) vs. 1.90 (±1.34)). Consistent to the previous
studies of the low-dose bumetanide for ASD, the side effects were rarely
reported (Tables S2 and S3). No significant difference in the cytokine levels
between the children with and without the gastrointestinal problems at the
baseline (Table S4). A
number of cytokine levels were changed significantly after the treatment of
bumetanide, but none of them was changed significantly after the treatment of
placebo (Table S6). No significant pairwise association could be
identified in the Discovery Set, the Validation Set and the Control Set among
four groups of variables, including the baseline CARS total score, the baseline
cytokine levels, the change of CARS total score, and the changes of cytokine
levels (Fig. S2).
In this study, we
observed a significant improvement of clinical symptoms with bumetanide
treatment in children with ASD, and such improvement was associated with a
pattern of changes in three cytokine levels, namely the IFN-γ, MIG and
IFN-α2 (r = 0.459 in the Discovery Set and r = 0.316
in the Validation Set). These cytokine levels at the baseline could improve the
prediction of the bumetanide responders compared with using the behavioural
assessments alone, and the best predictor achieved an AUC of 0.83 in the
independent test data set (Table S8). The implications of these findings may be twofold: (1) a significant
part of the clinical heterogeneity in the treatment effect of bumetanide for
ASD is associated with the differences in the immune system of patients, and
(2) the component score of cytokines had a potential to construct a blood
signature for predicting and monitoring the bumetanide treatment in young
children with ASD.
Accumulating
evidences support that IFN-γ can inhibit chloride secretion [38] and down-regulate both
the NKCC1 expression [16, 38] and the Na+-K+-ATPase
expression [16], which had been
implicated in the GABAergic dysfunction in ASD [10, 39].
The
cytokine-symptom association was identified in the changes after the treatment
of bumetanide but not before the treatment, suggesting that bumetanide might
interact with the cytokines and the changes of which contributed to the
treatment effect of bumetanide. Animal studies showed a rapid brain efflux of bumetanide,
but a number of clinical trials have shown a significant treatment effect for
neuropsychiatric disorders, including ASD, epilepsy and depression [41, 42]. These findings may suggest the
possible systemic effects of bumetanide as a neuromodulator for these
neuropsychiatric disorders. Considering its molecular structure, bumetanide has
been recently identified by an in vitro screen of small molecules that can act
as an anti-proinflammatory drug via interleukin inhibition [43]. This
anti-proinflammatory activity of bumetanide might alter the blood levels of
cytokines outside the brain-blood-barrier (BBB).
Our findings
may suggest that the identified canonical score of cytokines had a potential to
construct a blood signature for predicting and monitoring the bumetanide
treatment in young children with ASD. Accurately identifying patients who are
likely to respond positively to bumetanide can facilitate the precision
medicine for ASD. Our prediction model based on the cytokine levels before the
treatment may provide a potentially new tool for the precision medicine of
ASD.
In summary, we identified an association between the changes of
the cytokine levels and the improvements in symptoms after the bumetanide
treatment in young children with ASD, and found that the treatment effect of
bumetanide can be better characterised by an immuno-behavioural covariation.
This finding may provide new clinically important evidence supporting the
hypothesis that immune responses may interact with the mechanism of bumetanide
to restore the GABAergic function in ASD. This finding may also have relevance
for determining enriched samples of ASD children to participate in novel drug
treatment studies of drugs with a similar mode of action to bumetanide, but
with potentially greater efficacy and fewer side effects.
Conclusion
I think we can give the Shanghai
researchers 10 out of 10 for their paper.
Monty, aged 18 with ASD, has been to
Shanghai twice. It is a vast city, but well worth a visit. With the high speed
train network it is now very easy to travel around China, quite different to
when I visited as a teenager.
Hopefully the Chinese will continue in
their pursuit of precision medicine for autism. They do not have much
competition.
My perspective is a little different
because I know that a bumetanide responder can cease to be a responder when
affected by an inflammatory condition like allergy, which increases pro-inflammatory
cytokines like IL-6. This suggests that some people with elevated cytokines are
potential responders, you just have to use an anti-inflammatory therapy before
you start bumetanide therapy. The inflammatory cytokines shift the balance
between NKCC1 and KCC2 towards NKCC1 and so increasing intracellular
chloride. We also know that some people
need a dose higher than 0.5mg twice a day to see a large benefit; I have been
using 2mg once a day for several years.
The Chinese researchers have
established biomarkers for who is likely now to respond to bumetanide. This
certainly is a big step forward, if it can be replicated. This is not the same
as identifying who could respond to bumetanide, if their current inflammatory
condition was moderated. The levels of specific cytokines might indeed mark
someone as both a current non-responder, but also as a potential future
responder.
Autism is all about n=1, it is about
the exceptions being more important than the average.
Unlike the Shanghai researchers, I do not
really see Bumetanide as an anti-inflammatory therapy in my son’s Polypill, but
I do have therapies included that are.
Understanding inflammation will be a
key to treating autism using precision medicine. That is less simple that it sounds. When it
comes to preventing autism, inflammation in the mother is a key part of the
equation. This also gets complicated, maternal antibodies damage the brain of
the fetus, no genetic mutations were needed.

